Example of DynDom run
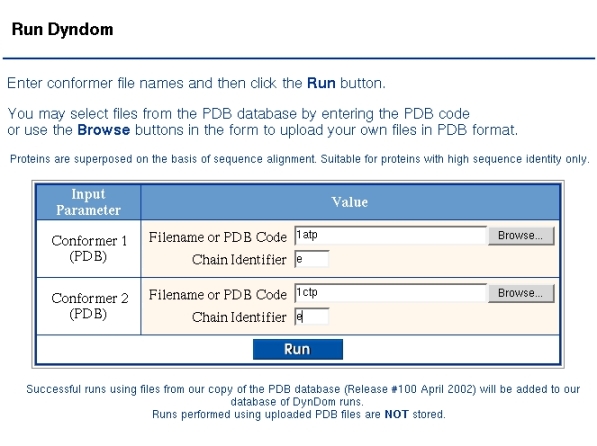
Below a run is described on cAMP protein kinase. Figure 1 shows the "Run DynDom" form used to identify the two conformations. As DynDom runs only on single chains the PDB code and chain identifier must be given for both conformers. If there is no chain identifier then the corresponding field is left blank. If the two chains come from the same PDB file then the same PDB code must be given in both fields.
Output from the DynDom program is shown in Figure 2. It shows the number of residues used for the analysis and the backbone root mean-square deviation (RMSD) from a least-squares superposition of the two proteins. For the program to proceed past this stage this RMSD must be larger than 0.1A. The subsequent output shows the output from the clustering process. Sometimes a single cluster can be split up into a number of domains, although in this case this has not happened. For each domain pair the ratio of interdomain to intradomain displacement is output. In this case the value is 1.44. This is greater than the minimum value which is set at 1.0 and so this pair will be accepted for the next stage of the analysis, which is the determination of the interdomain screw axis and the bending regions. Before this happens the program continues the clustering process but the third cluster is smaller than the minimum domain size which is set at 20 residues and the clustering process stops. The program then goes on to analyse the interdomain motion for the pair of domains found when there were just two clusters. The program chooses the largest domain as the fixed domain and the final output is the RMSD between the two conformations of this domain.
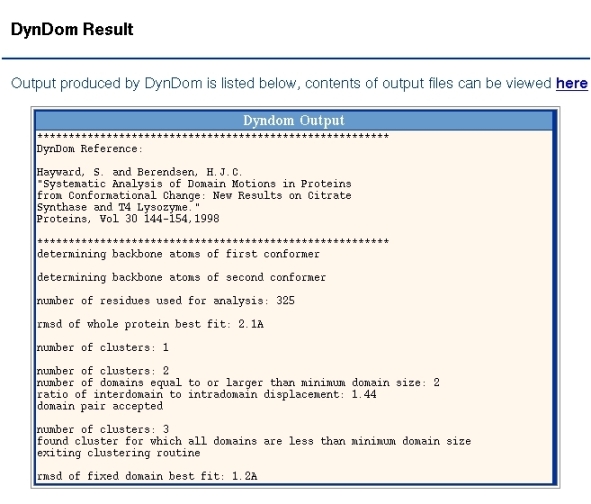
Figure 3 shows the parameters used in the DynDom run. The user of the server cannot set the window length, minimum ratio and minimum domain size. The minimum domain size and ratio, which refers to the ratio of interdomain to intradomain displacement, are fixed at 20 residues and 1.0, respectively, for all runs and do not change during the course of a run. The window length is set initially at 5 residues but if there is a null result it will automatically increase in two residue increments until a successful domain decomposition results or the window length exceeds 15 residues. A failed run means that there is no domain pair for which the domain sizes are greater than 20 residue and their ratio of interdomain to intradomain displacement is greater than 1.0. These criteria are set to ensure a consistency in the results.
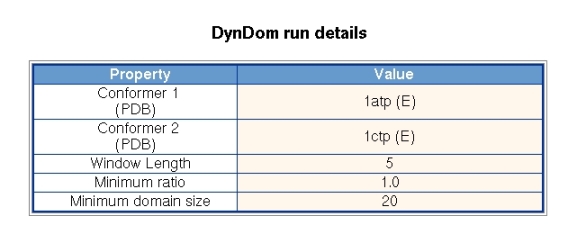
Figure 4 shows the domains section, where the segments that comprise a domain are given. The residue numbers are taken from the first PDB file of conformer 1. The numbering in the second conformer may by different. Each domain is given an identifying number. Domain number 1 comprises 223 residues and the backbone RMSD between the two conformations is 1.2A. Domain 2 comprises 98 residues and has an RMSD of 1.3A.
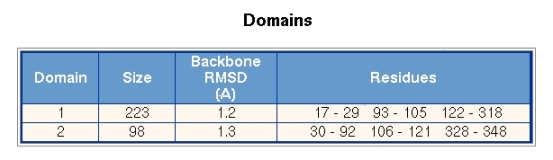
Figure 5 shows a pairwise alignment of both sequences colored according to domain or bending region. The numbering is taken from their respective PDB files and the sequences themselves come from the ATOM record not the SEQRES record of the PDB file.

Figure 6 details the interdomain motion. The fixed domain is identified as domain 1 in the "Domains" section and the moving domain as domain 2. The viewpoint is looking down the axis which is located where the lines cross. At the website a "mouseover", "mouseoff" movement will swap images of the two conformations allowing one to see the domain movement.
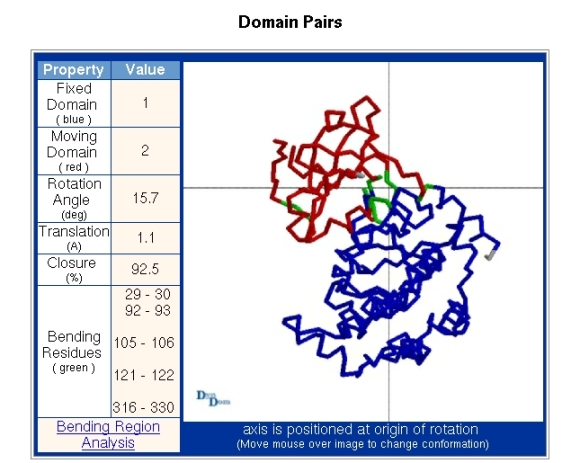
Figure 7 shows the section for downloading a script file for viewing locally using RasMol. The script contains the coordinates of the two conformations as well as an arrow which depicts the interdomain screw axis. The domain and bending regions of the first conformer will be colored accordingly, the second conformer being left uncolored.

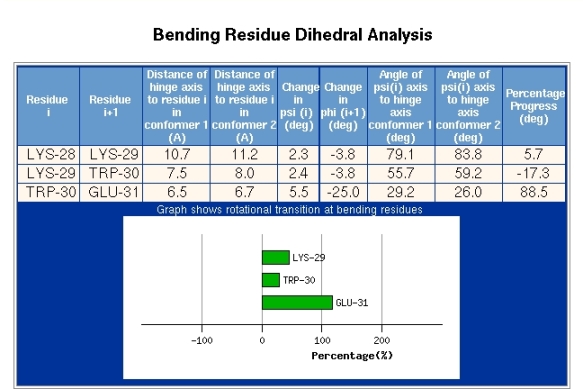
The section depicted in Figure 8 is accessed via the "Bending Region Analysis" link seen in the "Domain Pairs" section of Figure 6. The information depicted in Figure 8 is given for each bending segment. Figure 8 shows the result for the bending region extending from residue 29-30. The first two columns of data show the distance of the alpha-carbon atom of residue i from the interdomain screw axis for both conformations. The next two columns present the change in the psi-dihedral for residue i, and the change in the phi-dihedral of residue i+1, between the two conformations. The following columns give the angle between the psi-dihedral axis and the interdomain screw axis in both conformations. The final column is explained by first explaining the meaning of the plot. Bending regions are where the transition occurs between the two rotational properties of a pair of domains. The plot in Figure 8 is meant to reveal to which domain the residue indicated is dynamically closest. In the plot 0% indicates that the residue is rotating with the domain from which it originates which in this case is domain 1, as residue 29 is part of that domain (see Figure 4). 100% indicates that the residue is rotating with the domain in which the bending residue segment terminates which is domain 2, as residue 30 belongs to domain 2 (see Figure 4). These percentages are calculated by evaluating the degree to which a residue is rotating with the rotating domain (2 in this case) and is determined by calculating the rotation of the tetrahedron formed from the atoms N,C-alpha,C-beta, and C and comparing it to the rotation of domain 2. In this case residue 29 has a value of 50% which indicates that dynamically it is in between the two domains. Glu31 however has a value of about 125% which means it is rotating in the same direction as domain 2 relative to domain 1 but rotates a little more than the overall rotation of domain 2. In any case, this means that Glu31 is certainly closer associated with domain 2 than domain 1. The transition is seen to occur mainly between Trp30 and Glu31. The phi-dihedral of Glu31 changes by -25 degrees compared to 5.5 degrees for Trp30 indicating that is it the phi-dihedral of Glu31 that contributes most to the interdomain rotation. The final column in Figure 8 titled "Percentage Progress" gives the difference in the percentages of the corresponding two residues shown in the plot. The data presented in Figure 8 can be used to identify residues that are classic hinge-bending residues. Often there is considerable noise in this data and for interdomain rotations less than 20 degrees it may not be very informative.